We are interested in the design of proteins and cellular systems.
Proteins function in the context of a cell, and interact both physically (through binding) and biochemically (by exchanging substrates and creating post-translational modifications). These interactions provide the foundation for cells to maintain metabolic homeostasis and respond appropriately to environmental change. A need to maintain those interactions necessary to organismal fitness places constraints on protein structure, function, abundance and ultimately sequence. We aim to quantify these constraints and use this information to design new proteins, custom regulation, and robust synthetic cellular systems.
Our work makes use of both computation and experiment, primarily we work in E. coli as a model organism. Past work has involved statistical analysis of protein sequences, bacterial comparative genomics, machine learning, CRISPR-based tools for modulating gene expression, directed evolution, large-scale mutagenesis, and high-throughput assays of bacterial growth. Here we organize our research into three broad conceptual areas.
1. Quantifying and modeling the relationship between gene expression, environment, and growth.
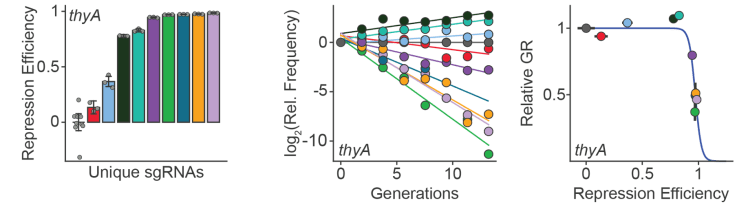
We are broadly interested to understand the evolutionary constraints on gene expression and protein abundance. How much of a given protein is enough? Or too much? Predicting how changes in gene expression modulate cell growth is critical to interpret the effects of disease-associated mutations and engineer new synthetic systems. To address this, we developed new CRISPR-based tools for titrating gene expression in E coli. We coupled these tools with high-throughput (sequencing based) growth rate measurements in custom-built turbidostats enabling precise environmental control. The resulting data is used to train and test predictive models. Going forward, we will apply these methods to optimize synthetic biochemical pathways, understand how environment impacts antibiotic sensitivity, and quantify constraints on enzyme expression genome wide.
Related publications:
R.M. Otto, A. Turska-Nowak, P.M. Brown, K.A. Reynolds (2024). A continuous epistasis model for predicting growth rate given combinatorial variation in gene expression and environment. Cell Systems. 15:134. [ paper ] [ code ]
A.D. Mathis, R.M. Otto, K.A. Reynolds (2020). A simplified strategy for titrating gene expression reveals new relationships between genotype, environment, and bacterial growth. Nucleic Acids Research , gkaa1073. [ paper ] [ code ]
2. Biochemistry in vivo: Understanding constraints on enzyme activity, stability, abundance, and sequence.
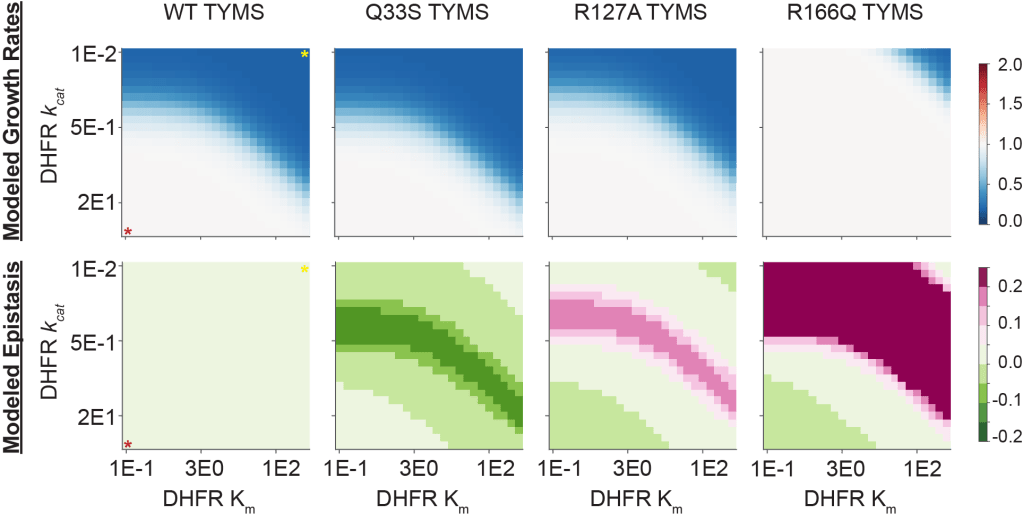
Proteins are often considered the building blocks of the cell – but their “design specifications” remain unclear. For a given enzyme, can we put bounds on the values for abundance, catalytic activity and stability sufficient to sustain metabolic pathway function and growth? A priori, it is not obvious if the bounds on these values are narrow or broad, to what extent the bounds are shaped by cellular context, and how these constraints are encoded in the protein sequence. In prior work, we used deep mutational scanning to investigate how intracellular protease effects constraints on protein activity and stability. We are also quantifying interactions between metabolic enzymes: for example, if one enzyme in a pathway acquires a loss of function mutation, how does it impact the selection pressure on downstream enzymes? In the future, we plan to develop new methods for contextually aware enzyme design –engineering of catalytic systems in a way that accounts for surrounding pathway reactions and constraints introduced by cellular context.
Related publications:
T. Nguyen, C. Ingle, S. Thompson and K.A. Reynolds (2024). The genetic landscape of a metabolic interaction. Nature Communications, 15:3351. [ paper ] [ code ]
S. Thompson, Y. Zhang, C. Ingle, K.A. Reynolds, and T. Kortemme (2020). Altered expression of a quality control protease in E. coli reshapes the in vivo mutational landscape of a model enzyme. eLife, 9:e53476. [ paper ] [ code ]
A.F. Schober, C. Ingle, J.O. Park, L. Chen, J.D. Rabinowitz, I. Junier, O. Rivoire, and K.A. Reynolds (2019). A two-enzyme adaptive unit in bacterial folate metabolism. Cell Reports, 27:3359-3379. [ paper ]
3. Evolution and engineering of allostery
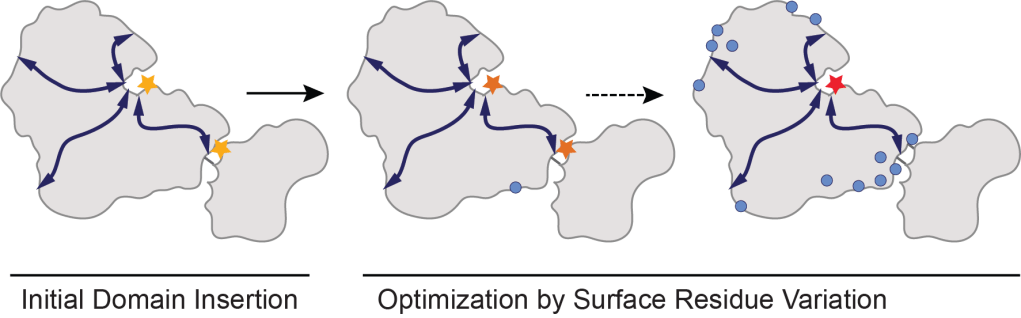
Allosteric regulation is a common feature of proteins in which a site distal to the active site can regulate protein activity. It is fast, reversible, and can be mediated by many different signals, including light, voltage, small molecules, and protein binding. An ability to predict allosteric surfaces and engineer new regulation has immense practical utility for the design of biosensors and discovery of new (allosterically-acting) therapeutics. In prior work, we showed that conserved protein surface sites that co-evolve with the active site can be used to engineer new allostery by domain insertion and post-translational modification. These “allosteric hotspots” seem to have latent allosteric potential that can readily be harnessed to generate new regulation. We are now developing new approaches to evolve allostery in the lab, with an eye to understanding how regulation is optimized. We are also performing biophysical experiments and MD simulations to understand what physical properties might distinguish allosteric hotspots.
Related publications:
J.W. McCormick, J.C. Dinan, M.A.X. Russo, K.A. Reynolds (2024). Local disorder is associated with enhanced catalysis in an engineered photoswitch. preprint. [bioRxiv]
J.W. McCormick, M.A.X. Russo, S. Thompson, A. Blevins, K.A. Reynolds (2021). Structurally distributed surface sites tune allosteric regulation. Elife (10): e68346. [paper] [ bioRxiv ] [ code ] [ talk ]
D. Pincus, J. Pandey, P. Creixell, O. Resnekov, and K.A. Reynolds (2018). Engineering allosteric regulation in protein kinases. Science Signaling (11):555. [paper][code].
K.A. Reynolds, R.N.McLaughlin, R. Ranganathan (2011). Hotspots for allosteric regulation on protein surfaces. Cell (147): 1564-1575. [paper]
